
You can read my previous posts on this drug here (
1,
2).
The Research: Part 2
The second study published on the efficacy of agomelatine was by Kennedy and Emsley (2006,
3).
This was a 6-week, double-blind, randomized, placebo-controlled study involving 212 patients. Dosage ranged from 25-50mg/day (dose adjustment at week 2 for poor responders). No other active comparator (e.g., paroxetine) was used in this study. Similar to the previous study (Loo et al, 2002), the efficacy of agomelatine on a severely depressed subpopulation was examine too.
Surprise, surprise, agomelatine was shown to be superior to placebo (HAM-D total score 14.1 +/- 7.7 versus 16.5+/- 7.4). Plot twist:
"The proportion of patients who were in remission by the end of the acute treatment period was not statistically different between the two treatment groups." Of course, that could be due to the short duration (6-weeks) of the study.
Remember this quote from the previous study I reviewed:
"25mg of agomelatine was significantly better than placebo at 2 weeks..., whereas this significant advantage for paroxetine...did not emerge until 4 weeks." Here is the survival analysis for this study:
The difference did not occur until week 4, the same as paroxetine in the previous study. So this study failed to replicate the result of the first study.
Common side-effects reported include:
"dizziness, nasopharyngitis and influenza were more common in the agomelatine group that placebo." Again, no sexual side-effects were reported (sorry, no fancy chart to show).
Part 3:
The third published study was by Olie and Kasper (2005,
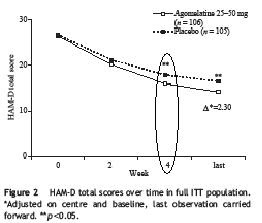
4). This study is similar in design as the study mentioned-above. At the end of 6-weeks, there was a superior response for agomelatine compared to placebo (3.44 point difference).
Here is the survival analysis curve for time to first response:

Here, you can see a difference was noted at week 2 (replicating the original result), but then they merge at week 4 (difference was still significant) and then separate again thereafter. What is interesting about placebo temporarily merging with the active drug at week four, is that there was a dose adjustment from 25mg to 50mg for poor responders at week 2. Probably not the robust result they were looking for, but a reaction non-the-less.

Reported side-effects are similar to the previous studies:
 Comment:
Comment: Both of these study are extremely short (6-weeks). 2/3 of depressed patients usually do not respond to their first anti-depressant. Moreover, while response rates (50% reduction in symptoms) are usually robust, remissions rates a paltry (usually 1/5-1/3 remission). No long-term information can be gathered from these two short-term studies. There is long-term data, but it's unpublished.
Side-effect do appear mild. However, many SSRI antidepressant trials show mild side-effects. It's not until the drug is widely prescribe do common side-effects become evident.
All three studies were biased against placebo (i.e., 1 week placebo wash-out period).
Keep in mind that these are published studies of positive trials. There are negative trials that are simply not published (I'm shocked!).
The European Medicines Agency, the parallel to the FDA, initially rejected the drug in 2006 (
5).
Here is what they said:

In case you cannot read the image, it says,
"The major concern of the CHMP was that the effectiveness of Valdoxan/Thymanax had not been sufficiently shown. The long-term study (the unpublished data I mentioned) did not show that the medicine was effective. The short-term studies shown that the medicine has an effect, but the extent of this did not allow the Committee to draw a firm conclusion on the medicine's effectiveness."The drug was finally approved in 2008 (
6). In their report they list all the submitted trials.


Some highlights:
-In study CL3-22, which included a fluoxetine comparator. This study, which was a short-term with a long-term (1 year) extension found that both agomelatine and fluoxetine were not statistically superior to placebo. (oops!).
-In study CL3-23 agomelatine and paroxetine were not statistically superior to placebo over the short-and-long term. (whoops!).
-CL3-24, the results were identical to CL3-33. (strike three, you're out!).
-Study CL3-21 was a relapse prevention study against placebo. At the end of the trial, agomelatine had a relapse rate of 26% versus 24% for the placebo group (strike four! wait that's not right). They did a
post-hoc analysis (i.e., statistical masturbation) and found that only for severely depressed patients there was a statistical difference. The proper thing to do at this point is to run a NEW study to test that intriguing hypothesis since the analysis was done after the fact. (It didn't happen, obviously).
-Efficacy in the elderly was not demonstrated
-Because of concerns over liver toxicity, liver monitoring is required. (do they require that for SSRI's)
Versus other AntidepressantsMuch of the hoopla around this drug has been it's supposed superiority against fluoxetine (Prozac). If you head over to the official website, they tout the findings of a recent study (
7). But is it really superior? The data submitted to the EMEA showed agomelatine to be equal to SSRI's (2 paroxetine studies, 2 fluoxetine studies, & 2 venlafaxine studies). With the exception of one study where superiority to sertraline (submitted later) was shown. Here's is what the EMEA had to say about the matter:
 "magnitude appears less than the active comparators."
"magnitude appears less than the active comparators."So that's 2 studies out of 8 that showed a superior effect. There are many studies in the literature that show one antidepressant being superior to another (
8). However, results like theses are the exception, not the rule.
The HypeBased on my review of the data, I'm not seeing much in the way of a wonderful new addition to the anti-depressant family. Aside from liver toxicity, side-effect profile does seem favorable, which is certainly an advantage compared to SSRI's. However, efficacy does not appear any greater than currently available treatments (maybe less effective overall). Just like SSRI's, there are a number of negative trials, so the effect is certainly not consistent.
Furthermore, during my review, I found 6 review articles (see my first post), which rehash the same 3 primary studies over and over again. What's worse, these 6 articles were published within a 3 year period and all in the journals for which Montgomery is the editor. They also read like the democratic party's "talking points" on health care reform, meaning, they all stay on message. That message being
"need for better antidepressants" "safety and tolerability" "unique mechanism of action." This strikes me as familiar to the recent trend in second generation antipsychotic articles (
9,
10,
11). What I truly enjoyed, though, is the SSRI bashing that was going on in these studies. Last Psychiatrist discussed quite well last year (
12,
13).
My Final VerdictSlightly better side-effect profile, actual clinical efficacy is uncertain.
 KENNEDY, S., & EMSLEY, R. (2006). Placebo-controlled trial of agomelatine in the treatment of major depressive disorder European Neuropsychopharmacology, 16 (2), 93-100 DOI: 10.1016/j.euroneuro.2005.09.002Pierre Olié, J., & Kasper, S. (2007). Efficacy of agomelatine, a MT1/MT2 receptor agonist with 5-HT2C antagonistic properties, in major depressive disorder The International Journal of Neuropsychopharmacology, 10 (05) DOI: 10.1017/S1461145707007766
KENNEDY, S., & EMSLEY, R. (2006). Placebo-controlled trial of agomelatine in the treatment of major depressive disorder European Neuropsychopharmacology, 16 (2), 93-100 DOI: 10.1016/j.euroneuro.2005.09.002Pierre Olié, J., & Kasper, S. (2007). Efficacy of agomelatine, a MT1/MT2 receptor agonist with 5-HT2C antagonistic properties, in major depressive disorder The International Journal of Neuropsychopharmacology, 10 (05) DOI: 10.1017/S1461145707007766




 In an attempt to counter his image as a professional douche bag, Mr. West "got into the Christmas spirit by making a surprise appearance at the Los Angeles Mission Saturday (Dec. 26). The Grammy winner, alongside longtime girlfriend Amber Rose, joined other volunteers and served lunch to the homeless, who greeted the Chicago native and took pictures. 'It's just important [to give back] when you're very blessed.'" (1).
In an attempt to counter his image as a professional douche bag, Mr. West "got into the Christmas spirit by making a surprise appearance at the Los Angeles Mission Saturday (Dec. 26). The Grammy winner, alongside longtime girlfriend Amber Rose, joined other volunteers and served lunch to the homeless, who greeted the Chicago native and took pictures. 'It's just important [to give back] when you're very blessed.'" (1).
































